

時間:

作者:保敏敏、呂蓓蓓、魏文芝、張敏娟
目的 建立往復筒溶出方法考察奧卡西平刻痕片仿制藥與原研藥分割后半片制劑的體外溶出行為相似性,評價原研藥和仿制藥的分劑量藥學特性差異。
方法 以pH1.2鹽酸溶液、pH4.5醋酸鹽緩沖液、pH6.8磷酸鹽緩沖液和水(均含0.5%十二烷基硫酸鈉 250 mL)為溶出介質,往復頻率為10dip·min–1,采用往復筒溶出裝置測定仿制藥和原研藥的溶出曲線,結合相似因子(f2)法評價仿制藥和原研藥的溶出行為相似性,并與槳法進行比較。采用脆碎度檢測儀及電子天平,通過人工掰分法和切藥器法測定各廠家半片制劑的脆碎度、分割后質量差異及質量損失。
結果 仿制藥A在4種溶出介質中的 f2 均>50,與原研藥的溶出行為相似;仿制藥B在4種溶出介質中f2均<50,和原研藥的溶出行為不相似。仿制藥A、B分割后質量差異、質量損失和脆碎度均高于原研藥。
結論 采用往復筒法測定奧卡西平半片制劑的溶出曲線相較于槳法具有良好的區分力,且仿制藥的分劑量質量控制相較于原研藥仍有一定差距。

奧卡西平是一種新型抗癲癇藥物,可用于治療成人和兒童癲癇的簡單或復雜部分性發作、全身強直陣攣發作等,酶誘導作用弱,不良反應少。目前,奧卡西平上市劑型主要為刻痕片,可掰分后使用,便于靈活調整劑量,方便用藥人群,降低治療成本。刻痕片的大小、硬度和刻痕線的形狀、深淺等因素會對分劑量的準確性造成影響,進而出現劑量偏小致使療效減弱或偏大而引起不良反應的問題。尤其對于老年人、兒童等特殊患者群體和治療窗窄的藥物,需要格外關注分劑量用藥的安全性。目前國內對于刻痕片多探討該劑型在臨床使用時分劑量的均勻性,相關深入評價研究較少,對于內在質量參數考察不足,中國藥典 2020年版尚未收錄相關通則。
往復筒法作為近年得到較快發展的新型溶出度檢查方法,已被各國藥典收載為標準方法,但國內相關實驗研究較少。該裝置主要通過將樣品放置在玻璃往復筒中,控制往復筒在溶出杯中的往復運動來測定藥物溶出量,具有更接近人體胃腸道蠕動生理環境的運行條件,能夠良好地反映體內外相關性。研究表明往復筒法對于部分普通片劑能夠實現和籃法或槳法類似的流體動力學特點,提供區分力良好的溶出曲線,具有評價溶出行為一致性的潛在優勢。
目前各國藥典標準所收載奧卡西平片溶出度檢查方法均為槳法,以含 0.6%十二烷基硫酸鈉(sodium dodecyl sulfate,SDS)水溶液作為溶出介質。本研究采用往復筒法測定奧卡西平半片制劑的溶出曲線,并與槳法進行比較,得到更具有區分力的溶出曲線,考察進口原研藥與國產仿制藥半片制劑的溶出行為差異。同時根據美國食品藥品監督管理局(FDA)及國家藥品監督管理總局藥品審評中心發布的仿制藥功能性刻痕相關研究指導原則,對奧卡西平片進行分割后質量差異、質量損失、脆碎度參數的考察,為國內奧卡西平片仿制藥一致性評價研究工作和處方工藝開發提供參考。
2.1 儀器
HPLC儀(Agilent1260型,G7111A型四元泵,G7129A 型自動進樣器,G7116A 型柱溫箱,G71157型二極管陣列檢測器)、往復筒溶出儀(推薦使用華溶DS-BIOAT往復筒法溶出系統);真空脫氣儀(推薦使用華溶DGU-900在線溶媒脫氣機);PB-21型酸度計、CP225D 型十萬分之一電子天平均購自德國Sartorious 公司;FT-2000型脆碎度檢測儀(天津市矽新科技有限公司);KQ-500DE型數控超聲波清洗器(昆山市超聲儀器有限公司)。
2.2 試藥與試劑
奧卡西平對照品(中國食品藥品檢定研究院,批號:100657-201102;純度:99.8%);奧卡西平片原研制劑(瑞士Novartis公司,規格:300mg;批號:TT434、TV182、TU897);奧卡西平片仿制制劑(國內A企業,規格:300mg;批號:18108、19003、19410,仿制藥 A),奧卡西平片仿制制劑(國內B企業,規格:300mg;批號:11805、11057、19102,仿制藥 B);SDS(SIGMA-ALDRICH,批號:SLBZ9853;含量≥99.0%);SDS(福晨化學試劑有限公司,批號:20190220;含量≥59.0%);SDS(國藥集團化學試劑有限公司,批號:F20090326;含量≥86.0%);乙腈為色譜純,其余試劑均為分析純,水為超純水。
3.1 色譜條件
色譜柱:ZORBAX SB C18(150mm×4.6mm , 5 μm);流動相:乙腈-0.05mol·L–1磷酸二氫鉀溶液(含0.2%三乙胺,用磷酸調節 pH至6.0)(35∶65);檢測波長:256nm ;流速:1.0mL·min–1;柱溫:30 ℃; 進樣量:10μL。
3.2 溶液的制備
3.2.1 對照品溶液的制備
精密稱取奧卡西平對照品約50mg,置50mL量瓶中,加流動相溶解并稀釋至刻度,作為對照品儲備液,精密量取2mL, 置10mL量瓶中,用流動相溶解并稀釋至刻度,搖勻,制成濃度約為 200μg·mL–1的對照品溶液。
3.2.2 供試品儲備液的制備
精密稱取奧卡西平片 ( 批號 :18108)細粉適量(約相當于奧卡西平50mg),置50mL量瓶中,加流動相溶解并稀釋成每1mL約含1mg奧卡西平的供試品儲備液。
3.2.3 供試品溶液的制備
精密稱取奧卡西平片 ( 批號:18108) 細粉適量 (約相當于奧卡西平10mg),置50mL量瓶中,加流動相適量,超聲使奧卡西平溶解并定量稀釋至刻度, 搖勻,濾過,取續濾液,即得。
3.2.4 陰性樣品溶液的制備
按奧卡西平片處方比例制成缺奧卡西平的空白輔料樣品,稱取適量,加流動相溶解,定量稀釋并濾過,得到陰性樣品溶液。
3.3 方法學驗證
3.3.1 專屬性試驗
分別取“2.2”項下對照品溶 液、供試品溶液和陰性樣品溶液于“2.1”項下色譜條件進樣測定,記錄色譜圖,見圖1,結果表明輔料對主峰測定無干擾。
3.3.2 線性關系考察
分別精密移取“2.2.1”項下 對照品儲備液 0.5 ,1,2,4,8 mL ,置10 mL量瓶中,用流動相稀釋至刻度,搖勻,取上述溶液及對 照品儲備液按“2.1”項下色譜條件進行測定。以色譜峰面積(Y)與對應的質量濃度(X)進行線性回歸,得回歸方程 Y=16 349X+64.506(r=0.999 9)。結果表明奧卡西平在 49.9~998.0 μg·mL–1 內與峰面積之間 線性關系良好。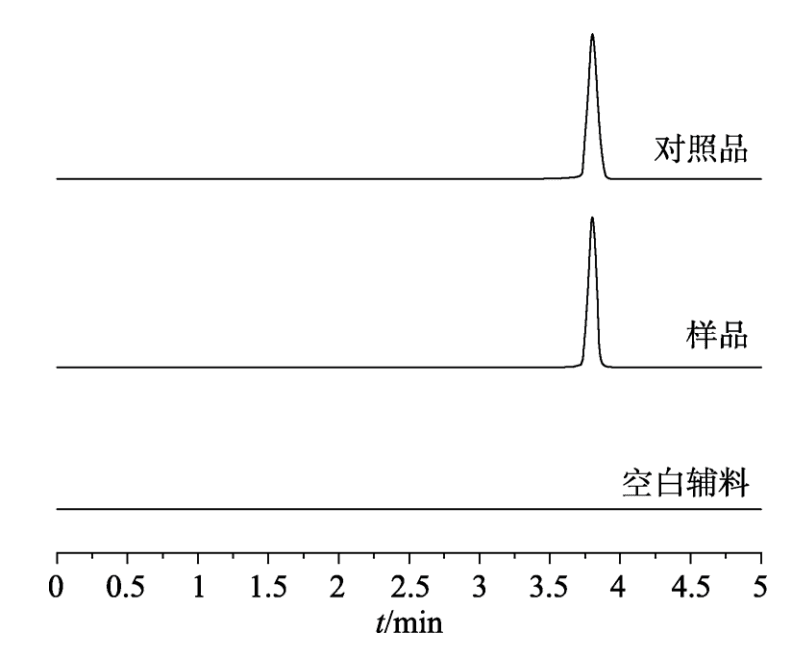
3.3.3 儀器精密度試驗
取“2.2.1”項下奧卡西 平對照品溶液,按“2.1”項下色譜條件連續進樣 6 次, 記錄峰面積。結果顯示RSD為 0.1%(n=6),本方法儀器精密度良好。
取“2.2.1”項下奧卡西 平對照品溶液,按“2.1”項下色譜條件連續進樣 6 次, 記錄峰面積。結果顯示RSD為 0.1%(n=6),本方法儀器精密度良好。
3.3.4 重復性試驗
取同一批奧卡西平片(批號:18108),按“2.2.3”項下方法平行制備供試品溶液6 份,按“2.1”項下色譜條件進行含量測定。測得奧卡西平含量的 RSD為1.2%(n=6),表明本方法重復性良好。
3.3.5 加樣回收率試驗
移取“2.2.2”項下供試 品儲備液2mL ,置10mL 量瓶中,共12份,再分別精密加入“2.2.1 ”項下對照品儲備液 0.4,2.8,4,5.2mL,各3份,用流動相稀釋至刻度,搖勻。按“2.1”項下色譜條件進樣測定,外標法計算回收率,結果見表1 ,表明方法回收率良好。
3.3.6 溶液穩定性試驗
取“2.2”項下對照品溶 液和供試品溶液, 室溫放置 0,2,6,12,18,24 h,按“2.1”項下色譜條件測定,記錄峰面積。奧卡 西平對照品和供試品峰面積的RSD分別為0.4%, 0.5%,表明該溶液穩定性良好。
3.3.7 耐用性試驗
取“2.2.3”項下供試品溶液, 在不同pH(5.8,6.0,6.2)、不同柱溫(25,30,35 ℃)以及不同流速(0.8,1.0,1.2mL·min–1)條件下按“2.1”項下色譜條件進樣測定。結果顯示pH、柱 溫、流速于一定范圍內變化時, 奧卡西平含量的RSD 均<2%,對測定結果無影響,表明該方法耐用性良好。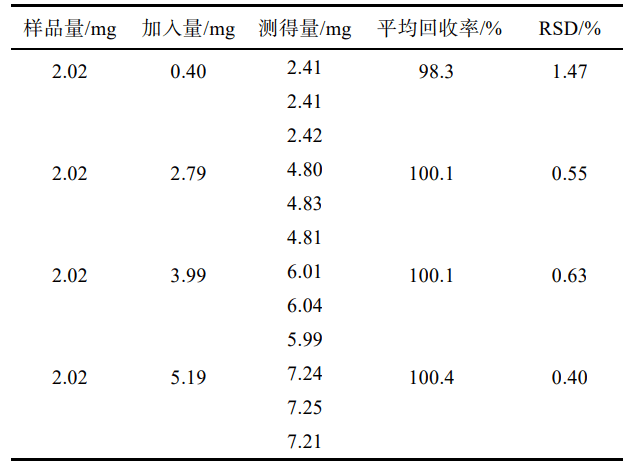
3.4 往復筒溶出方法的確定
3.4.1 SDS 濃度的考察
以水為溶出介質,分別測 定加入 SDS 0.3%,0.4%,0.5%和 0.6%時原研藥和 仿制藥的溶出度,比較不同SDS用量對奧卡西平半片的溶出影響,見表2。結果顯示,加入0.3%,0.4%SDS 時各廠家制劑累積釋放量均<80%。加入0.6%SDS時 2 種仿制藥的相似因子均>50,溶出條件寬松,無區分力。因此確定SDS的用量為0.5%。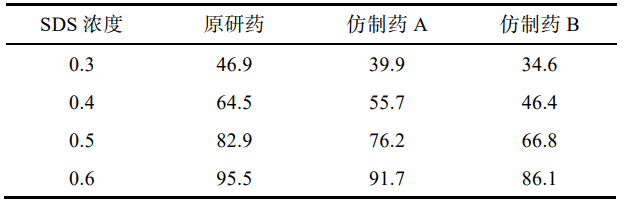
3.4.2 往復頻率的考察
以0.5%SDS水溶液250 mL為溶出介質,分別測定原研藥半片在往復速率為 5,10,20dip·min–1時的溶出度,結果見表3。不同往復頻率下原研藥的溶出度無顯著差異,溶出曲線基本一致,見圖2。往復頻率為10dip·min–1時起泡較少,故本研究選用10dip·min–1進行溶出度的測定。
3.4.3 SDS 來源的考察
選取進口以及2個國產廠家生產的SDS,以10dip·min–1測定原研藥在 0.5%SDS 水溶液中的溶出度,分別于5,10,15,20,30,45min取樣1.5mL,按“2.1 ”項下色譜條件測定。

結果顯示,使用SIGMA- ALDRICH、福晨和國藥生產的SDS得到的樣品10 min 時的溶出度RSD分別為2.1%,11.8%和7.5%。SIGMA-ALDRICH生產的SDS測得的樣品批內均一性良好,選擇該廠家SDS作為溶出度試驗的增溶劑。見表4。

3.5 溶出曲線測定
3.5.1槳法
參照美國藥典(USP43)中對于150mg 規格的奧卡西平片采用的溶出度測定方法,取本品6 片,以掰分后半片作為溶出樣品,轉速60r·min–1,溫度37℃,溶出體積900mL,分別在含0.3%SDS 的 pH1.2 鹽酸溶液、pH4.5醋酸鹽緩沖液、pH6.8磷酸鹽緩沖液和水中測定 溶出度,于5 ,10,15,20,30,45min時取溶液適量,0.45μm微孔濾膜濾過,作為供試品溶液。另精密稱定奧卡西平對照品適量,加流動相定量稀釋成質量濃度約為200μg·mL–1的溶液,作為對 照品溶液進樣。按“2.1”項下色譜條件測定,外標法計算不同時間點各廠家半片的溶出量,繪制溶出曲線,結果見圖3。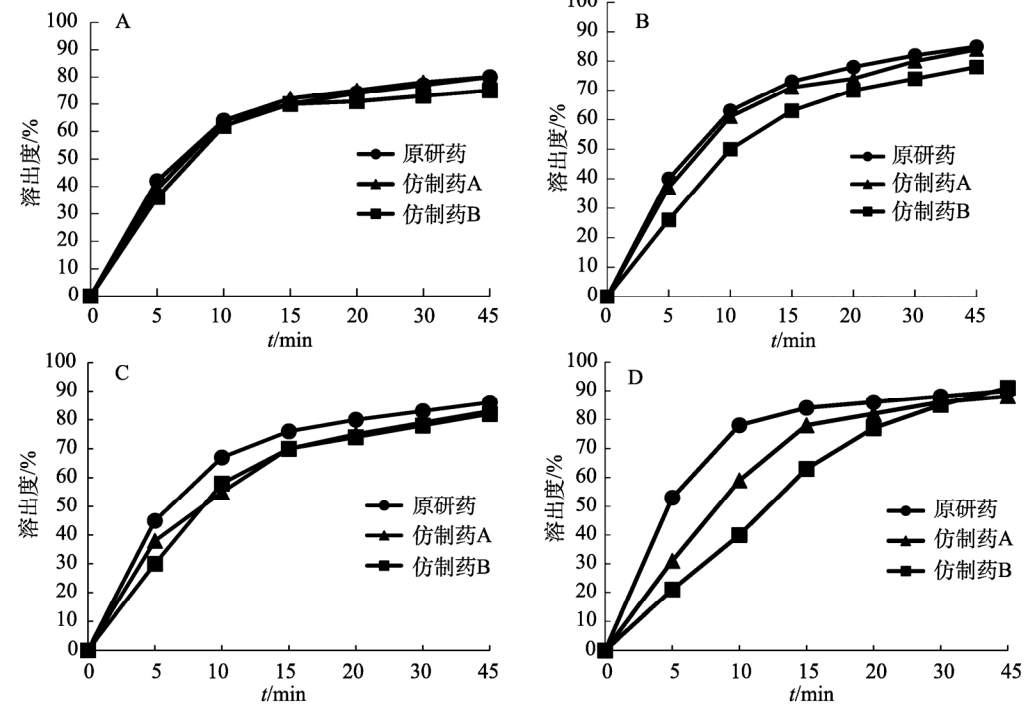
3.5.2 往復筒法
取本品6片,以掰分后半片作為溶出樣品,采用往復筒溶出裝置的單排管進行溶出試驗,設定往復速率10dip·min–1;不加上篩網,下篩網選擇100目,材質為聚丙烯酰胺;水浴溫度37 ℃ ; 分別以 0.5% SDS水溶液、0.5% SDS鹽酸溶液 (pH1.2) 、 0.5% SDS醋酸鹽緩沖液(pH 4.5) 、0.5%SDS 磷酸鹽緩沖液(pH 6.8)為溶出介質,體積為250mL;分別在 5,10,15,20,30,45min 時取樣1.5mL,用 0.45μm微孔濾膜濾過,作為供試品溶液。另精密稱定奧卡西平對照品適量,加流動相定量稀釋成質量濃度約為600μg·mL–1的溶液,作為對照品溶液進樣。按 “2.1”項下色譜條件測定,外標法計算不同時間點各廠家半片的溶出量, 繪制溶出曲線,結果見圖4。
3.6 相似性評價
根據《普通口服制劑溶出度試驗技術指導原則》,通過計算原研制劑與仿制制劑在不同溶出介質中的相似因子(f2)評價溶出曲線相似性,結果見表5~6。往復筒法相較于槳法,所得到的溶出曲線 具有更強的區分力。
3.7 質量差異

隨機選取本品30 片,以人工掰分和切藥器2種不同的方式分割成2個半片, 取每片中1個分割后部分進行稱重,比較其與平均半片的質量差異。分割后部分質量超出平均半片質量85%~115%的不能>1個,如果有>1個分割部分超過上述標準,或者有1個分割部分>75%~125%的限度,則不符合要求。結果顯示,原研藥通過2種分割方法得出的半片質量均一性均良好,未出現不合格半片;仿制藥A經切藥器法得到的半片質量差異大且出現1個不合格半片;仿制藥 B人工掰分和切藥器法各出現1個不合格半片。半片質量計算結果見表 7。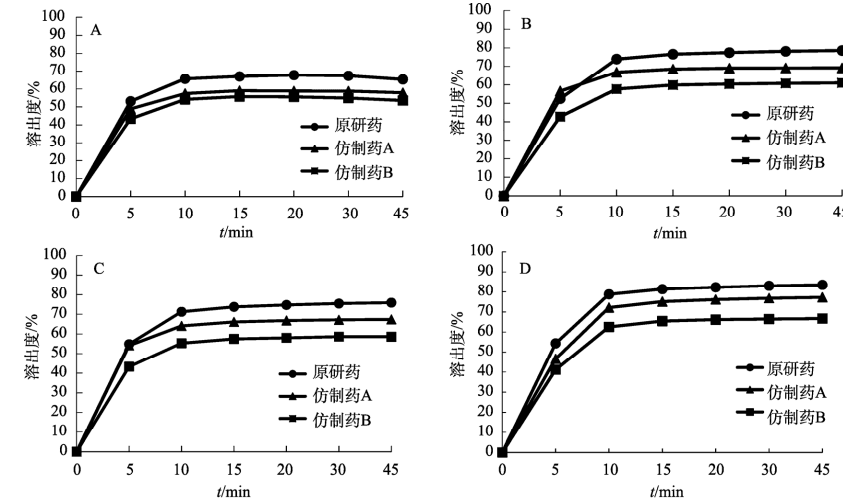

3.8 分割質量損失
用手掰和切藥器法各自分割15整片并精密稱定,和分割前質量比較,計算分割后整片的質量損失。結果顯示,原研藥的分割質量損失低于仿制藥,仿制藥A在切藥器法中質量損失最大,仿制藥B通過人工掰分質量損失最多,見表 8。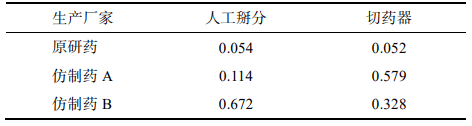
3.9 脆碎度
根據中國藥典2020年版通則0923,采用脆碎度檢測儀對原研藥和仿制藥分割后半片進行檢查,質量減失均<1%,且無斷裂、龜裂及粉碎片。2 種分割方式下原研藥的脆碎度均低于仿制藥。仿制藥B通過人工掰分法得到的半片脆碎度最高。見表 9。
4.1 色譜條件的選擇
采用中國藥典 2020年版奧卡西平片含量測定 方法時發現主成分峰前出現一個分離度差的色譜峰,影響含量測定的準確性,經與對照品色譜圖比較,排除其為雜質峰的可能。考慮奧卡西平結構中存在酰胺基,pH3.0的流動相可提供H+ ,酸 催化條件下奧卡西平易發生酰胺-亞胺醇式異構化反應,出現2種異構體的動態平衡,可能是導致該 HPLC 方法色譜圖出現無法分離的兩色譜峰的原因。參考美國藥典(USP43)方法將pH調至6.0,并測試不同品牌的C18柱系統適用性,結果顯示奧卡西平主峰前后均未出現分裂峰,主峰峰形良好。因此本研究采用調整后的色譜方法進行奧卡西平半片的含量測定。
4.2 溶出裝置的選擇
本研究采用往復筒法,在4種溶出介質中均得到比槳法更有區分力的溶出曲線,結合相似因子說明仿制藥A與B之間、仿制藥與原研藥之間的溶出行為均存在顯著性差異。采用該方法可以進一步評價產品內在品質,從而為改進仿制藥生產工藝提供數據參考。
4.3 篩網規格
往復筒溶出裝置的上下篩網規格一般為8~400 目,本研究通過試驗不同目數的篩網發現上篩網添加與否對藥物在往復筒中的溶解無明顯影響。對下篩網選用40,78, 100目進行考察,發現目數為100目時能夠防止樣品粉末漏出往復筒,避免溶出損失,故最終確定不加上篩網,下篩網為100目,可滿足實驗要求。
4.4 可分割性差異
仿制藥A的外觀和原研藥類似,為淺黃色膠囊形片, 雙面中刻痕。在使用切藥器時,由于其質地比較堅硬,切割阻力大易導致不規則崩裂現象,繼而出現分割后半片不均一、分割質量損失高的情況。仿制藥B為圓形片,單面中刻痕且刻痕較淺。其片形使得徒手掰分比較費力,容易出 現質量不均一的半片,導致分割后片劑的質量損失較仿制藥A更為顯著。因此,仿制藥的刻痕功能在分割劑量方面不及原研藥。
本研究采用往復筒法比較國產與進口奧卡西 平半片制劑的體外溶出行為,在4種溶出介質中獲得的溶出曲線均能有效區分不同廠家的奧卡西 平片。結果顯示國產仿制藥的溶出行為與進口原研藥有不同程度的差異。同時通過考察分割質量損失等刻痕片的關鍵質量屬性,提示國產仿制制 劑分劑量質量控制存在提升空間。
略
如需原文,請聯系小編






