

時間:

作者:劉德鵬,李倩,郝貴周,丁靜雯,張貴民
摘 要
目的 建立一種有區(qū)分力的流通池法檢查泊沙康唑口服混懸液的體外溶出。
方法 以研磨法制成不同粒度分布的泊沙康唑混懸液中間體,并通過調(diào)節(jié)輔料黃原膠的加入量,制備成 具有不同粒度分布和不同黏度的泊沙康唑口服混懸液。對關(guān)鍵參數(shù)(玻璃珠用量、加樣方式以 及流速)進(jìn)行篩選,建立檢查泊沙康唑口服混懸液體外溶出的流通池法。用新建立的流通池法 和 FDA 溶出度數(shù)據(jù)庫收載的槳法分別檢測泊沙康唑口服混懸液的體外溶出曲線。
結(jié)果 槳法測得的溶出曲線藥物釋放較快,20 min 時已基本釋放完全;而新建立的流通池法測得的溶出曲 線藥物釋放緩慢,并且對混懸液的粒度分布和黏度差異顯示出良好的區(qū)分力。
結(jié)論 與槳法相比,流通池法檢查泊沙康唑口服混懸液的體外溶出時具有更高的區(qū)分力。
關(guān) 鍵 詞

01 簡 介
泊沙康唑(posaconazole)是一種三唑類抗真菌藥,具有抗菌譜廣、活性強(qiáng)、耐受性好、安全性高等特點,廣泛應(yīng)用于臨床,具有良好的治療效果。泊沙康唑被開發(fā)成多種劑型上市,其中,泊沙康唑口服混懸液(規(guī)格:40 mg·mL,商品名:諾科飛、Noxafil)由原先靈葆雅公司研發(fā),于2005年11月在歐洲上市,2013年6月在中國獲批上市,目前國內(nèi)尚無仿制產(chǎn)品上市。
流通池法作為一種新型的溶出度檢查方法,已收載于2020 年版《中國藥典》,但暫未見具體檢查品種實例。在該方法下,預(yù)先加熱至合適溫度的溶出介質(zhì)經(jīng)恒流泵以一定流速持續(xù)通過流通池,與流通池中的藥物接觸并使其釋放。可以通過改變?nèi)艹瞿J健⒄{(diào)節(jié)流速、更換流通池內(nèi)徑、調(diào)整玻璃珠用量等方式調(diào)整溶出速度,使溶出方法可以更好地模擬體內(nèi)環(huán)境。尤其是在閉環(huán)模式下,樣品時刻暴露于新鮮的溶出介質(zhì)中,始終維持著適宜的漏槽條件,更適用于難溶性藥物的體外溶出研究,在一些藥物的體外溶出研究中顯示出更好的區(qū)分力。
文獻(xiàn)研究顯示,在進(jìn)食高脂肪食物時,泊沙康唑的平均粒徑為1.7μm和2.3μm的混懸液在健康受試者體內(nèi)不具有生物等效性,大粒徑混懸液的相對生物利用度為小粒徑的 76%,表明原料的粒度分布是泊沙康唑口服混懸液的關(guān)鍵質(zhì)量屬性。但因其處方中含有二氧化鈦等水不溶性輔 料,采用常規(guī)粒徑分析方法檢測時易受干擾,難以準(zhǔn)確剖析參比制劑中原料的粒度分布。因此,本研究結(jié)合流通池法的特點,建立了一種能區(qū)分泊沙康唑口服混懸液中原料粒度分布差異的體外溶出檢測方法,為該產(chǎn)品的開發(fā)及質(zhì)量控制提供支持。
02 材料
2.1 儀器
DYNO-MILL型研磨機(jī)(瑞士WAB公司);L5M型高剪切乳化器(英國Silverson 公司);Mastersizer 3000 型激光粒度分析儀(英國Malvern公司);XS204型電子分析天平(瑞士 Mettler Toledo公司);MCR302 型流變儀(奧地利 Anton Paar 公 司);溶出儀(推薦使用華溶儀器DS-1406AT全自動取樣溶出系統(tǒng))、流通池法溶出儀(推薦使用華溶儀器DS-7CP PLUS 流池法溶出系統(tǒng));UltiMate 3000型高效液相色譜儀(美國 Thermo Fisher Scientific公司)。
2.2 試藥
泊沙康唑原料藥(含量:100.8%,批號:20210434)、泊沙康唑?qū)φ掌罚ê浚?9.8%,批號:RSP-0271-20201011,浙江奧翔藥業(yè)股份有限公司);聚山梨酯80(南京威爾藥業(yè)集團(tuán)股份有限公司);枸櫞酸鈉、二甲硅油、麥芽糖漿、十二烷基硫酸鈉(SDS)(湖南爾康制藥股份有限公司);枸櫞酸(湖南九典宏陽制藥有限公司);黃原膠(CP Kelco U.S.,Inc.);苯甲酸鈉(南京化學(xué)試劑股份有限公司);甘油(豐益生物科技有限公司);香精(森馨香精色素科技中國有限公司);二氧化鈦(江蘇宏遠(yuǎn)藥業(yè)有限公司);純化水(山東新時代藥業(yè)有限公司)。
03 方法與結(jié)果
3.1 泊沙康唑口服混懸液的制備
取聚山梨酯808.7g,加至275g中純化水,攪拌至溶解,加入泊沙康唑原料藥35g,繼續(xù)攪拌至分散均勻;用高剪切乳化器乳化(10000 r·min)10min;再將乳化后的混懸液經(jīng)研磨機(jī)濕法研磨(轉(zhuǎn)速8.0m·s ),進(jìn)一步減小粒徑,循環(huán)研磨2次收集混懸液1;同法制備,循環(huán)研磨3次收集混懸液2。采用激光粒度分析儀濕法模塊檢測粒度分布,遮光度10%~20%、顆粒折射率1.657、吸收率0.01、攪拌速度2000r·min,測量時間30s,測定3次取平均值。結(jié)果顯示:混懸液1的粒度分布D10為(0.92±0.015)μm;D50為(2.15±0.051)μm;D90為(9.78±0.022)μm;混懸液2的粒度分布D10 為(0.56±0.017)μm;D50為(1.67±0.048)μm;D90為(6.82±0.021)μm。
依次取二甲硅油2.6g、黃原膠2.62g、苯甲酸鈉1.75 g、甘油87.25g、香精4.4g、二氧化鈦3.5g、麥芽糖漿350g,分別加至275g純化水中,攪拌至溶解分散均勻后,用枸櫞酸-枸 櫞酸鈉緩沖液(pH 4.0)調(diào)節(jié)pH至4.0~5.0,得溶液1;另取黃原膠2.22g,其他同法制備,得溶液2。
將上述混懸液與溶液按表1方式以1∶1混合均勻,即得泊沙康唑口服混懸液。分別測定沉降體積比,結(jié)果顯示,BHXY1、BHXY2和BHXY3的沉降體積比均為1.0。用流變儀分別測定黏度,測定溫度25℃,轉(zhuǎn)子 CP50,結(jié)果顯示,BHXY1、BHXY2和BHXY3的黏度分別為(356±45)mPa·s、(212±32)mPa·s 和(353±30)mPa·s。
3.2 HPLC 方法及其驗證
采用 HPLC測定泊沙康唑口服混懸液的溶出度,并對分析方法進(jìn)行驗證。
表 1 泊沙康唑口服混懸液的組成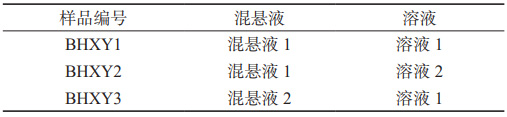
3.2.1 色譜條件
色譜柱Diamonsil C18(250mm×4.6mm,5μm);流動相乙腈-水(60∶40);流速1.0 mL·min;檢測波長262nm;柱溫25℃;進(jìn)樣量10μL。
3.2.2 專屬性
按照處方量配制空白輔料,取“2.4”項下各溶出介質(zhì)定量稀釋制成空白輔料溶液。取各溶出介質(zhì)及其配制的空白輔料溶液,進(jìn)樣測定,結(jié)果顯示輔料和溶出介質(zhì)對主成分的測定均無干擾,該方法專屬性較好。
3.2.3 線性關(guān)系考察
精密稱取泊沙康唑?qū)φ掌芳s200mg,置100mL量瓶中,用流動相溶解并稀釋至刻度,搖勻,作為對照品儲備液。分別取對照品儲 備液適量,定量稀釋制成每1mL 中含泊沙康唑約10、50、80、100、150、200 和 250 μg 的溶液, 作為系列線性溶液,進(jìn)樣測定,以峰面積(A)對濃度(C)進(jìn)行線性回歸,回歸方程為:A = 104.41C-142.63,r=0.9993,表明泊沙康唑在10~250μg·mL 內(nèi)與峰面積的線性關(guān)系良好。
3.2.4 精密度
取“3.2.3”項下的100μg·mL的對照品溶液,連續(xù)進(jìn)樣測定6次,結(jié)果峰面積的RSD為0.62%,表明方法精密度良好。
3.2.5 回收試驗
按處方量配制輔料溶液,精密量取適量置50mL量瓶中,再分別加入“3.2.3”項下配制的泊沙康唑?qū)φ掌穬湟?2、5、6mL(各 3 份),加0.3%SDS 溶液稀釋至刻度,搖勻,過濾。進(jìn)樣檢測,并計算回收率,結(jié)果泊沙康唑低、中、 高濃度樣品的平均回收率為99.54%、99.63%和99.62%,RSD為0.37%、0.36%和0.17%(n=3)。
3.2.6 耐用性
分別采用不同色譜柱、柱溫(20、25和30℃)及流動相比例進(jìn)行耐用性試驗,結(jié)果各條件下均能滿足系統(tǒng)適用性要求,說明方法的耐用性良好。
3.3 流通池體外溶出檢測方法的研究
流通池法有兩種運行模式,即開環(huán)模式和閉環(huán)模式,示意圖見圖1。開環(huán)模式下新鮮的溶出介質(zhì) 連續(xù)泵入樣品池中,是非循環(huán)模式;閉環(huán)模式下溶 出介質(zhì)在泵的作用下重復(fù)進(jìn)入樣品池中,是循環(huán)模式。相比而言,開環(huán)模式可以使樣品在良好的漏槽 條件下持續(xù)釋放,因此本研究選用開環(huán)模式。
3.3.1 流通池法初步試驗
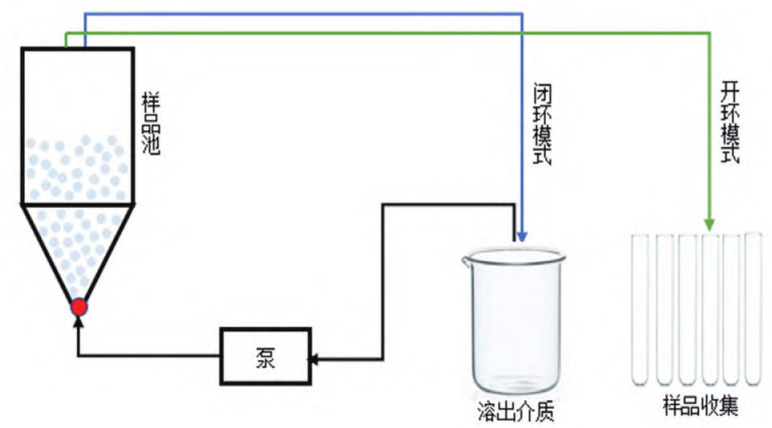
圖 1 流通池開環(huán)與閉環(huán)模式示意圖
選用內(nèi)徑為22.6mm的標(biāo)準(zhǔn)流通池,在倒置的錐體下端放入直徑為5mm 的紅寶石以防止樣品池中的介質(zhì)倒流入管路,用注射器吸取5mL樣品并精密稱定,加至樣品池錐體下端紅寶石上,樣品池內(nèi)充填直徑1mm的玻璃 珠 20g,濾室內(nèi)加入GF/D濾膜,將濾室與樣品池組裝好后,裝入溶出儀,開環(huán)模式,溫度37℃,流速8 mL·min,分別于10、20、30、45、60、90、120、180、240和360min時取樣,經(jīng)0.45μm水相微孔濾膜過濾,取濾液作為供試品溶液;另精密稱取泊沙康唑?qū)φ掌愤m量,用流動相溶解并 稀釋制成100μg·mL的對照品溶液。取供試品溶液和對照品溶液分別進(jìn)樣測定,采用外標(biāo)法計算藥物濃度并繪制累計溶出曲線。
3.3.2 方法參數(shù)優(yōu)化
選取含0.3%SDS 的水溶液 為溶出介質(zhì),使用BHXY3 樣品對流通池法的關(guān)鍵參數(shù)如玻璃珠用量、加樣方式及流速等進(jìn)行篩選和優(yōu)化。
① 玻璃珠用量:用注射器取5mL泊沙康唑口服混懸液,加至樣品池的錐體下端,再分別加入10、15和20g玻璃珠,流速為8mL·min,結(jié)果溶出曲線(見圖2)。玻璃珠用量對溶出速度基本無影響,但較多量的玻璃珠可以獲得更好的分散效果,因此玻璃珠用量選擇20 g。

圖 2 玻璃珠用量對溶出速度的影響(n = 6)
② 樣品加樣方式:用注射器取5mL泊沙康唑口服混懸液,分別在樣品池錐形下端、玻璃珠上層以及與玻璃珠交替分散加樣(見圖 3),玻璃珠用量為20g,流速為8mL·min,溶出曲線(見圖4)。

圖 3 樣品池中加樣方式示意圖
A. 在樣品池錐形下端加入
B. 與玻璃珠交替分散加入
C. 在玻璃珠上層加入
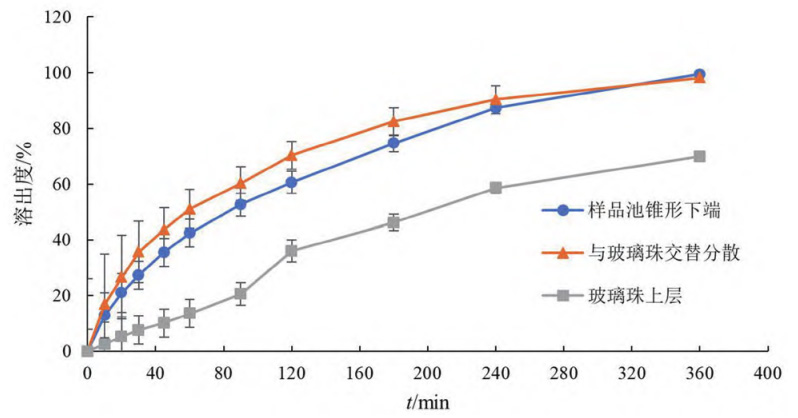
圖 4 加樣方式對溶出速度的影響(n = 6)
結(jié)果表明,在樣品池錐形下端先加入混懸液后再加入玻璃珠,玻璃珠在重力作用下沉至紅寶石處,使混懸液均勻分散至玻璃珠層,可使藥物緩慢釋放,且重復(fù)性好;混懸液與玻璃珠交替加入樣品池時,玻璃珠與混懸液間易形成斷層,且操作煩瑣,不同人員間操作差異較大;混懸液加至玻璃珠上層時,由于混懸液黏度較大,難以分散至玻璃珠層,而集中于玻璃珠層表面,使藥物的釋放緩慢。因此,選用在樣品池錐形下端加入樣品。
③ 流速:用注射器取5mL泊沙康唑口服混懸液加至樣品池內(nèi),再加入20g 玻璃珠,流速分別為4、8、16 mL·min,溶出曲線(見圖 5)。
結(jié)果表明,混懸液的溶出速率與流速成正相關(guān),流速越大,溶出越快;溶出達(dá)85%以上時,流速為4、8、16 mL·min時所需的溶出時間分別為360min以上、240min和90min。為保證方法對粒度分布和黏度有一定區(qū)分力,并且在6h內(nèi)可溶出完全,流速選用8mL·min 。

圖 5 流速對溶出速度的影響(n = 6)
3.4 流通池法和槳法的對比
泊沙康唑?qū)儆贐CSⅡ類藥物,在酸性介質(zhì)中溶解度較高,在弱酸性和中性介質(zhì)中的溶解度較低。因此,將不同pH的溶出介質(zhì)選定為:pH1.0鹽酸溶液、含0.3%SDS 的 pH 4.5 醋酸鹽緩沖液、含0.3%SDS 的pH 6.8磷酸鹽緩沖液和含0.3%SDS的水溶液。使用新建立的流通池法檢測 BHXY1、BHXY2和BHXY3在以上四種介質(zhì)中的溶出曲線,并與槳法進(jìn)行對比。
FDA 溶出度方法數(shù)據(jù)庫中收載的泊沙康唑口 服混懸液的溶出度測定方法為槳法,轉(zhuǎn)速為25 r·min,介質(zhì)為含 0.3%SDS的溶液,介質(zhì)體積為900mL。參照該方法,按以下方式投樣檢測:用注射器吸取5mL混懸液并精密稱定,迅速按壓注射器投樣并開啟溶出儀計時,分別于10、20、30、45、60min時取樣,用0.45μm濾膜過濾,續(xù)濾液作為供試品溶液;對照品溶液配制同“3.3.1”項下。取供試品溶液和對照品溶液分別進(jìn)樣測定,采用外標(biāo)法計算藥物濃度并繪制溶出曲線(見圖 6)。
結(jié)果表明,采用槳法檢測的泊沙康唑口服混懸 液溶出均較快,20min時已基本溶出完全,對處方中粒度分布和黏度差異無區(qū)分力;而采用流通池法檢測時,混懸液在四種溶出介質(zhì)中緩慢溶出,且對處方中粒度分布和黏度差異具有較好的區(qū)分力。
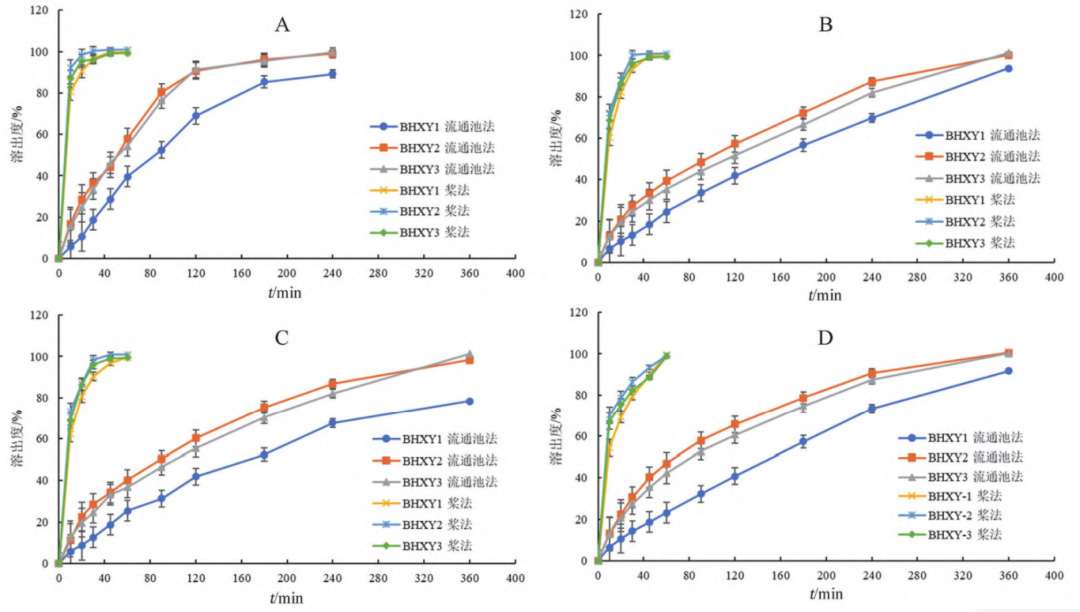
圖 6 兩種方法測得泊沙康唑口服混懸液在四種介質(zhì)中的溶出曲線(n = 6)
04 討 論
本研究采用研磨法制備了兩種不同粒度分布 的粗混懸液,中位值粒徑分別為2.15μm和1.67μm,與文獻(xiàn)報道的原研制劑臨床用樣品的粒徑中位值相當(dāng),并通過調(diào)節(jié)助懸劑黃原膠的加入量制備出不同黏度的泊沙康唑口服混懸液。其中BHXY1與BHXY2為大粒徑產(chǎn)品(2.15μm)、BHXY3為小粒徑產(chǎn)品(1.67μm),BHXY2黏度較小。槳法檢測時,用注射器將混懸液加入溶出杯中后,混懸液快速分散,啟動攪拌槳后帶動混懸液進(jìn)一步分散。由于藥物粒徑較小呈現(xiàn)出快速溶出,對混懸液粒徑的微小差異不具有區(qū)分力,與文獻(xiàn)報道一致,相反,用新建立的流通池法檢測時,混懸液呈現(xiàn)出相對緩慢的溶出,且能區(qū)分處方中粒度分布和黏度的微小差異。
綜上,本研究建立的流通池法檢測泊沙康唑口服混懸液體外溶出的方法具有重現(xiàn)性好、區(qū)分力強(qiáng)的優(yōu)點,可為該產(chǎn)品的開發(fā)提供更好的指示,也為其他難溶性混懸制劑的體外溶出研究提供了參考。
05 參考文獻(xiàn)
略






